 2024, Vol. 41
2024, Vol. 41






2. 上海中化科技有限公司, 上海 200120
2. Shanghai Sinochem Technology Co. Ltd., Shanghai 200120, China
烯烃复分解反应是指在过渡金属(钼、钨和钌等)催化下,底物烯烃的碳-碳双键被切断并重组的过程[1-3]。作为合成碳-碳双键的有效方法之一,烯烃复分解反应已被用于药物、生物大分子及高分子聚合物的合成过程中[4-6]。由于此反应条件温和、产率较高、对底物官能团的耐受性较强(羟基、羧基和羰基等),从而受到研究者的广泛关注[7]。
烯烃复分解反应最初是用高氧化态的钼和钨等配合物进行催化[8],但由于反应条件苛刻,催化剂对空气及水分敏感,所以其应用受到了限制[9]。1992年,Grubbs等[10]首次合成了结构明确的钌复分解催化剂1(图 1),并用于降冰片烯的开环聚合反应。1996年,基于膦配体的钌卡宾烯烃复分解催化剂2(Grubbs-Ⅰ代)被合成,其在空气中稳定,对底物的官能团适用性强[11]。与膦配体相比,氮杂环卡宾(NHC)具有更强的给电子能力,可与过渡金属Ru之间形成较强的相互作用力,从而避免反应过程中金属配合物的快速分解[12],因此将催化剂2中的1个膦配体用氮杂环卡宾替代,可得到稳定性更强、催化活性更高的Grubbs-Ⅱ代催化剂3[13]。当催化剂3中的膦配体被吡啶取代时,可得到Grubbs-Ⅲ代催化剂4[14],其可用于催化开环交叉聚合反应,以此来制备具有低分散相对分子质量的聚合物[15-17]。

|
| 图 1 Grubbs等合成的钌复分解催化剂 Fig.1 Grubbs olefin metathesis catalysts |
| |
观察催化剂的发展进程,其中配体的改变对催化剂的活性和引发速率有显著影响,这也指明了催化剂的改进以及创新方向[18]。Lynn等[19]使用Grubbs-Ⅰ代的前驱体,在其结构中引入含季铵盐的膦配体,合成了可溶于水且稳定性良好的催化剂5和6,用于催化水相中环状烯烃的开环聚合反应。Stewart等[20]在催化剂7的基础上,使其和1, 2-二乙烯基苯反应,以88%的产率得到了催化剂8。2018年Grubbs等在Grubbs-Ⅱ代催化剂中引入膦胺配体合成催化剂9,结果发现P—N键的引入加速了膦配体的解离,从而提高了催化剂的引发速率[21]。结构式见图 2。

|
| 图 2 催化剂5~9的结构式 Fig.2 Structures of Ru complexes 5—9 |
| |
在以上研究的基础上,本研究合成了3种新型的钌烯烃复分解催化剂10~12,并采用核磁共振波谱对其结构进行表征。首先测试了催化剂2、10~12在25 ℃下放置相同时间后的稳定性(以二苯甲酮为内标),其次以二烯丙基丙二酸二乙酯为模板底物,在30 ℃下使用核磁共振氢谱监测反应进程,测试了催化剂2,10~12的催化性能及引发速率,最后测试了催化剂对RCM反应和CM反应的催化活性。
1 实验部分 1.1 试剂与仪器2-氟苯甲醛,2-氯苯甲醛,2-溴苯甲醛,叔丁醇钾,三环己基膦(PCy3),氢化钙,二苯甲酮,分析纯,天津希恩思生化科技有限公司;二烯丙基丙二酸二乙酯,分析纯,上海阿拉丁;三苯基膦二氯化钌[RuCl2(PPh3)3],分析纯,天津凯美维德科技有限公司;对甲苯磺酰肼,甲醇(CH3OH),二缩三乙二醇,石油醚(PE),二氯甲烷(DCM),江天化工有限公司。
柱层析硅胶,高效薄层层析板,烟台新诺化工,200~300目;Bruker AV400型核磁共振波谱仪,德国Bruker公司。
1.2 实验过程 1.2.1 催化剂10~12的制备催化剂的合成路线如图 3所示。

|
| 图 3 催化剂合成路线10~12 Fig.3 Synthesis of Ru catalysters 10—12 |
| |
(1) 对甲苯磺酰腙的合成:在圆底烧瓶中加入对甲苯磺酰肼(3.72 g,20.0 mmol)和CH3OH (35 mL),搅拌溶解后,缓慢加入2-氟苯甲醛(2.48 g,20.0 mmol),反应混合物在室温下搅拌2 h后有白色固体析出,将固体抽滤并用CH3OH(3×20 mL)洗涤,得到2-氟苯甲醛对甲苯磺酰腙(4.68 g,产率:80%)。
(2) 重氮苄的合成:在圆底烧瓶中加入2-氟苯甲醛对甲苯磺酰腙(2.64 g,9.03 mmol)和二缩三乙二醇(30 mL),在60 ℃下加热搅拌使其溶解,之后缓慢加入叔丁醇钾(1.82 g,16.22 mmol),反应混合物在60 ℃下搅拌1 h,待溶液颜色变为红色后反应完成,将其冷却后倒入冰水中(70 mL),并用PE萃取(3×50 mL),合并有机相备用。
(3) 催化剂合成:氮气环境下,在圆底烧瓶中加入RuCl2(PPh3)3(2.50 g,2.6 mmol)和DCM(40 mL),搅拌溶解后,将反应体系降至-78 ℃,新制备的重氮苄的石油醚溶液逐滴滴加至上述钌配合物的溶液中,滴加完毕后在此温度下继续反应30 min。之后将反应混合物升至室温,氮气环境下向其中缓慢滴加PCy3(3.40 g,12.0 mmol)的DCM(20 mL)溶液,混合物颜色由黄绿色逐渐变为紫色,继续搅拌30 min。反应结束后减压除去部分溶剂,向剩余混合液中加入CH3OH(100 mL),可观察到有紫色固体析出,将固体抽滤并用CH3OH洗涤(3×20 mL),干燥之后得到催化剂10(1.71 g,产率78%)。催化剂11和12根据10的路线进行合成,分别得到催化剂11(1.67 g,产率:75%),催化剂12(1.78 g,产率:76%)。
1.2.2 催化剂的核磁共振波谱数据催化剂10: 1H NMR (CDCl3): δ 20.56 (s, Ru=CH), 9.07 (s, 1H), 7.65 (d, J=6.5 Hz, 1H), 7.06 (t, J=7.8 Hz, 1H), 6.96 (t, J=9.6 Hz, 1H), 2.61, 1.75, 1.44, and 1.21 [all m, P(C6H11)3]。13C NMR (101 MHz, CDCl3) δ 282.18, 152.75 (d, JCF=256.8 Hz), 141.20 (d, JCF=7.1 Hz), 133.98, 132.50 (d, JCF=8.8 Hz), 124.79 (d, JCF=3.7 Hz), 116.33 (d, JCF=23.4 Hz), 32.00 (t, J=9.2 Hz), 29.62, 27.83 (t, J=5.2 Hz), 26.51。31P NMR (162 MHz, CDCl3) δ 36.50 (s, PCy3)。19F NMR (376 MHz, CDCl3) δ -108.06。
催化剂11:1H NMR (400 MHz, CDCl3) δ 20.83 (s, Ru=CH), 9.27 (d, J=8.0 Hz, 1H), 7.56 (t, J=7.7 Hz, 1H), 7.32 (d, J=8.0 Hz, 1H), 7.19 (t, J=7.6 Hz, 1H), 2.58, 1.72, 1.44, and 1.21 [all m, P(C6H11)3]。13C NMR (101 MHz, CDCl3) δ 288.54, 148.85, 134.79, 131.27, 130.81, 130.65, 127.68, 32.05 (t, J=9.1 Hz), 29.68, 27.84 (t, J=5.3 Hz), 26.51。31P NMR (162 MHz, CDCl3) δ 36.59 (s, PCy3)。
催化剂12:1H NMR (400 MHz, CDCl3) δ 20.73 (s, 1H), 9.33 (d, J=8.0 Hz, 1H), 7.55 (d, J=8.0 Hz, 1H), 7.45 (t, J=7.6 Hz, 1H), 7.24 (d, J=15.3 Hz, 1H), 2.59 (d, J=12.7 Hz, 6H), 1.67, 1.44, and 1.22 [all m, P(C6H11)3]。13C NMR (101 MHz, CDCl3) δ 291.64, 149.79, 134.95, 134.06, 131.25, 128.40, 123.51, 32.08 (t, J=8.9 Hz), 29.72, 27.86, 26.52。31P NMR (162 MHz, CDCl3) δ 36.59 (s, PCy3)。
1.2.3 催化反应的一般过程关环复分解反应(Entries 1~11):在史莱克管中加入催化剂0.005 mmol,氮气环境下,加入底物(0.5 mmol)的DCM(0.5 mL)溶液,将其置于25 ℃下搅拌相应的时间。反应结束后,将反应混合物过硅胶柱,并用DCM淋洗,得到的混合物经过旋蒸浓缩后,快速分离提纯得到产物2a~2k。
交叉复分解反应(Entry 12 and 13):在史莱克管中加入催化剂0.012 5 mmol,氮气环境下,加入苯乙烯或4-氟苯乙烯(5.0 mmol)和苯基烯丙基醚(0.5 mmol)的DCM(1 mL)溶液,反应混合物在40 ℃下搅拌12 h。反应结束后,将反应混合物过硅胶柱,并用DCM淋洗,所得混合物经旋蒸浓缩,分离、纯化得到产物2l和2m。
2 结果与讨论 2.1 催化剂2,10~12的稳定性研究在核磁管中加入15 μmol的催化剂、0.6 mL CDCl3,以二苯甲酮(7.5 μmol)为内标,在25 ℃下放置相同时间,通过对δ=7.47~7.51 ppm(t)和δ=20.84 ppm(s)(以催化剂11为例)的峰面积进行积分比较(图 4),以此评估催化剂的稳定性[22]。结果发现随着时间变长,催化剂逐渐被分解,96 h后催化剂2几乎被完全分解,而催化剂10,11还保留有部分催化活性(图 5),根据Hong等[23]对催化剂分解过程的研究,发现催化剂上1个膦配体的解离,可能使卤素原子与钌卡宾之间形成配位作用,进而减慢催化剂的分解速度。

|
| 图 4 二苯甲酮为内标不同时间的催化剂11的1H NMR谱 Fig.4 1H NMR spectrum of the catalyst 11 with diphenylmethanone under different time |
| |

|
| 图 5 25 ℃下催化剂2、10~12在CDCl3中的稳定性,通过1H核磁共振波谱监测 Fig.5 Stability tests of complexes 2, 10~12 in CDCl3 at 25 ℃ monitored by 1H NMR spectroscopy |
| |
以二烯丙基丙二酸二乙酯为RCM反应的模板底物,对催化剂10~12的催化动力学进行分析,利用1H NMR监测反应进程。通过比较δ=2.61 ppm(dt)和δ=2.89 ppm (s)的峰面积,来判断产物1b的转化率[24],结果如图 6所示。催化剂10具有较快的引发速率,且随着催化反应进行,催化速率缓慢增加,40 min后转化率达到70%。催化剂11和12引发速率相差较小,但催化剂11只有55%的转化率。

|
| 图 6 催化剂2、10~12对产物1b的转化率,通过1H核磁共振波谱监测 Fig.6 Conversion to product 1b using catalysts 2, 10~12 monitored by 1H NMR spectroscopy |
| |
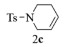
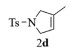
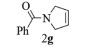
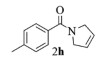
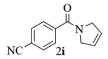
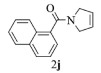
进一步探究了催化剂10~12对不同底物的催化性能,结果如表 1所示。对于N,N-二烯丙基对甲苯磺酰胺(Entry 1),催化产率可达到95%以上,当底物烯烃的链长增加时(Entry 2),催化反应仍能顺利进行。对于不对称取代的烯烃(Entry 3),催化活性虽有所降低,但仍可达到较高的产率(>85%);而当底物烯烃的双键上连接有取代基时,催化效率大幅下降(Entry 4),甚至不能进行催化反应(Entry 5),升高温度或增加催化剂的物质的量,都没有明显效果,说明双键上连有4个取代基时,底物烯烃的位阻会对催化剂的性能产生很大影响;对于烯炔类的底物催化剂10~12仍有较好的催化效果(Entry 6)。对于N,N-二烯丙基苯甲酰胺及其衍生物(Entries 7~11),催化剂10~12具有良好的官能团适用性,皆可催化生成相应的关环复分解产物,但对于—CN取代的N, N-二烯丙基苯甲酰胺,催化剂10~12只有60%的催化产率,其可能是因为—CN的强吸电子效应,对催化活性产生了影响。催化剂10~12同样可以用于催化苯乙烯与苯基烯丙基醚的CM反应,并以大约80%的产率得到产物2l和2m (Entry 12和13)。
| Entry | Substrate | Product | Yield |
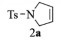
| 1 | 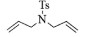 |
 |
10, 96% 11, 95% 12, 97% |
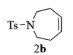
| 2 | 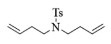 |
 |
10, 91% 11, 92% 12, 91% |
| 3 | 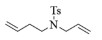 |
 |
10, 89% 11, 90% 12, 89% |
| 4 | 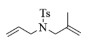 |
 |
10, 45% 11, 43% 12, 46% |
| 5 | 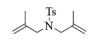 |
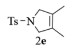 |
n.r. |
| 6b | 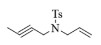 |
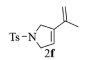 |
10, 89% 11, 91% 12, 92% |
| 7 | 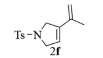 |
 |
10, 89% 11, 90% 12, 89% |
| 8 | 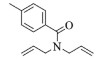 |
 |
10, 86% 11, 88% 12, 91% |
| 9 | 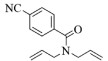 |
 |
10, 58% 11, 57% 12, 61% |
| 10 | 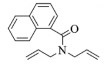 |
 |
10, 91% 11, 92% 12, 94% |
| 11 | 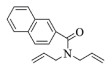 |
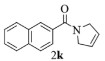 |
10, 89% 11, 88% 12, 91% |
| 12c |  |
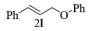 |
10, 78% 11, 81% 12, 82% |
| 13c | 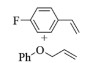 |
 |
10, 85% 11, 84% 12, 87% |
| a: The reaction was carried out at 25 ℃ for 1 h; b: The reaction was carried out at 25 ℃ for 24 h; c: The reaction was carried out at 40 ℃ for 12 h. Yield: isolated yield. Abbreviation: n.r.-no reaction. | |||
2a:1H NMR (400 MHz, CDCl3) δ 7.72 (d, J=8.3 Hz, 2H), 7.32 (d, J=8.0 Hz, 2H), 5.65 (s, 2H), 4.12 (s, 4H), 2.43 (s, 3H)。13C NMR (101 MHz, CDCl3) δ 143.45, 134.20, 129.78, 127.43, 125.46, 54.86, 21.56。
2b:1H NMR (400 MHz, CDCl3) δ 7.59 (d, J=7.9 Hz, 2H), 7.22 (d, J=8.0 Hz, 2H), 5.67 (t, J=3.5 Hz, 2H), 3.28-3.15 (m, 4H), 2.34 (s, 3H), 2.24 (d, J=5.2 Hz, 4H)。13C NMR (101 MHz, CDCl3) δ 143.11, 136.24, 130.23, 129.68, 127.05, 48.27, 29.90, 21.51。
2c:1H NMR (400 MHz, CDCl3) δ 7.58 (d, J=8.0 Hz, 2H), 7.23 (d, J=8.0 Hz, 2H), 5.66 (m, 1H), 5.56-5.47 (m, 1H), 3.48 (s, 2H), 3.08 (t, J=5.7 Hz, 2H), 2.33 (s, 3H), 2.11 (m, 2H)。13C NMR (101 MHz, CDCl3) δ 143.54, 133.26, 129.63, 127.64, 125.04, 122.72, 44.79, 42.65, 25.25, 21.50。
2d:1H NMR (400 MHz, CDCl3) δ 7.64 (d, J=7.9 Hz, 2H), 7.25 (d, J=7.9 Hz, 2H), 5.17 (s, 1H), 3.99 (s, 2H), 3.89 (s, 2H), 2.35 (s, 3H), 1.58 (s, 3H)。13C NMR (101 MHz, CDCl3) δ 143.35, 135.06, 134.29, 129.73, 127.44, 119.07, 57.68, 55.13, 21.52, 14.06。
2f:1H NMR (400 MHz, CDCl3) δ 7.74 (d, J=8.3 Hz, 2H), 7.33 (d, J=8.0 Hz, 2H), 5.59 (t, J=2.0 Hz, 1H), 4.98 (s, 1H), 4.77 (s, 1H), 4.23 (ddd, J=11.3, 5.7, 3.6 Hz, 4H), 2.42 (s, 3H), 1.85 (s, 3H)。13C NMR (101 MHz, CDCl3) δ 143.52, 139.18, 136.57, 133.99, 129.82, 127.46, 120.64, 114.60, 55.53, 54.37, 21.56, 20.00。
2g:1H NMR (400 MHz, CDCl3) δ 7.53 (m, 2H), 7.42 (m, 3H), 5.92 (m, 1H), 5.78~5.69 (m, 1H), 4.46 (m, 2H), 4.25~4.16 (m, 2H)。13C NMR (101 MHz, CDCl3) δ 169.77, 136.74, 129.82, 128.38, 128.33, 126.72, 125.78, 125.27, 55.74, 53.35。
2h:1H NMR (400 MHz, CDCl3) δ 7.33 (d, J=7.8 Hz, 2H), 7.10 (d, J=7.8 Hz, 2H), 5.87~5.68 (m, 1H), 5.68~5.54 (m, 1H), 4.34 (t, J=2.0 Hz, 2H), 4.24~4.00 (m, 2H), 2.27 (s, 3H)。13C NMR (101 MHz, CDCl3) δ 169.96, 139.98, 133.82, 128.93, 126.90, 125.86, 125.28, 55.81, 53.43, 21.39。
2i:1H NMR (400 MHz, CDCl3) δ 7.75 (d, J=8.3 Hz, 2H), 7.66 (d, J=8.2 Hz, 2H), 5.95 (m, 1H), 5.84~5.68 (m, 1H), 4.45 (dt, J=4.0, 1.8 Hz, 2H), 4.25~4.09 (m, 2H)。13C NMR (101 MHz, CDCl3) δ 167.73, 140.93, 132.38, 127.55, 125.87, 125.01, 118.13, 113.50, 55.58, 53.51。
2j:1H NMR (400 MHz, CDCl3) δ 7.83 (m, 3H), 7.56-7.40 (m, 4H), 5.85 (s, 1H), 5.59 (s, 1H), 4.55 (s, 2H), 3.84 (s, 2H)。13C NMR (101 MHz, CDCl3) δ 169.29, 135.20, 133.54, 129.29, 128.93, 128.47, 127.09, 126.40, 125.66, 125.45, 125.29, 124.78, 123.70, 54.95, 52.92。
2k:1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.91~7.77 (m, 3H), 7.60 (dd, J=8.4, 1.7 Hz, 1H), 7.55~7.41 (m, 2H), 5.89 (dt, J=6.8, 2.4 Hz, 1H), 5.77~5.67 (m, 1H), 4.49 (tt, J=4.1, 2.0 Hz, 2H), 4.21 (td, J=4.2, 2.3 Hz, 2H)。13C NMR (101 MHz, CDCl3) δ 169.96, 134.11, 133.80, 132.64, 128.50, 128.31, 127.80, 127.14, 126.71, 126.65, 126.00, 125.25, 124.12, 55.93, 53.55。
2l:1H NMR (400 MHz, CDCl3) δ 7.31 (ddt, J=27.5, 17.5, 8.0 Hz, 7H), 6.96 (d, J=7.9 Hz, 3H), 6.73 (d, J=16.0 Hz, 1H), 6.43 (dd, J=14.0, 7.5 Hz, 1H), 4.69 (d, J=5.8 Hz, 2H)。13C NMR (101 MHz, CDCl3) δ 158.66, 136.50, 133.01, 129.55, 128.64, 127.94, 126.63, 124.55, 120.95, 114.81, 68.58。
2m:1H NMR (400 MHz, CDCl3) δ 7.33 (dtd, J=27.1, 7.2, 6.4, 2.3 Hz, 4H), 7.11~6.87 (m, 5H), 6.68 (d, J=15.9 Hz, 1H), 6.33 (dt, J=16.0, 5.8 Hz, 1H), 4.67 (dd, J=5.8, 1.5 Hz, 2H)。13C NMR (101 MHz, CDCl3) δ 162.51 (d, JCF=247.2 Hz), 158.60, 132.65 (d, JCF=3.4 Hz), 131.84, 129.56, 128.16 (d, JCF=8.0 Hz), 124.26 (d, JCF=2.2 Hz), 120.99, 115.56 (d, JCF=21.6 Hz), 114.77, 68.44。19F NMR (376 MHz, CDCl3) δ 13.91。
3 结论合成了3种新型的苄亚基钌邻位含卤素的催化剂10~12,并通过核磁共振波谱确定了结构。通过比较3种催化剂的稳定性,发现与催化剂2相比,催化剂10~12的稳定性有所提升。以二烯丙基丙二酸二乙酯为RCM反应的模型底物,发现催化剂10具有较快的引发速率和良好的催化性能,在40 min后转化率达到70%。最后测试了催化剂对RCM反应和CM反应的催化活性,发现3种催化剂对不同底物均具有良好的催化活性和官能团适用性,但当双键上连有多个取代基时,会对RCM反应的催化性能产生影响。
| [1] |
杜创, 王春雨, 张国, 等. 降冰片烯开环易位聚合反应的分子量及分子量分布控制[J]. 高等学校化学学报, 2007, 28(10): 2018-2020. DU Chuang, WANG Chunyu, ZHANG Guo, et al. Molecular weight and molecular weight distribution control of ring-opening metathesis polymerization of norbornene[J]. Chemical Journal of Chinese Universities, 2007, 28(10): 2018-2020. (in Chinese) |
| [2] |
VOUGIOUKALAKIS G C, GRUBBS R H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts[J]. Chemical Reviews, 2010, 110(3): 1746-1787. DOI:10.1021/cr9002424 |
| [3] |
HOVEYDA A H, ZHUGRALIN A R. The remarkable metal-catalysed olefin metathesis reaction[J]. Nature, 2007, 450: 243-251. DOI:10.1038/nature06351 |
| [4] |
MORZYCKI J W. Application of olefin metathesis in the synthesis of steroids[J]. Steroids, 2011, 76(10/11): 949-966. |
| [5] |
MOL J C. Application of olefin metathesis in oleochemistry: An example of green chemistry[J]. Green Chemistry, 2002, 4(1): 5-13. DOI:10.1039/b109896a |
| [6] |
HERBERT M B, MARX V M, PEDERSON R L, et al. Concise syntheses of insect pheromones using Z-selective cross metathesis[J]. Angewandte Chemie (International Ed in English), 2013, 52(1): 310-314. DOI:10.1002/anie.201206079 |
| [7] |
TRNKA T M, GRUBBS R H. The development of L2X2RuCHR olefin metathesis catalysts: an organometallic success story[J]. Accounts of Chemical Research, 2001, 34(1): 18-29. DOI:10.1021/ar000114f |
| [8] |
SCHROCK R R, DEPUE R T, FELDMAN J, et al. Preparation and reactivity of several alkylidene complexes of the type W(CHR')(N-2, 6-C6H3-iso-Pr2)(OR)2 and related tungstacyclobutane complexes. Controlling metathesis activity through the choice of alkoxide ligand[J]. Journal of the American Chemical Society, 1988, 110(5): 1423-1435. DOI:10.1021/ja00213a014 |
| [9] |
SCHOETTEL G, KRESS J, OSBORN J A. A simple route to molybdenum-carbene catalysts for alkene metathesis[J]. J Chem Soc, Chem Commun, 1989(15): 1062-1063. DOI:10.1039/C39890001062 |
| [10] |
NGUYEN S T, JOHNSON L K, GRUBBS R H, et al. Ring-opening metathesis polymerization (ROMP) of norbornene by a Group Ⅷ carbene complex in protic media[J]. Journal of the American Chemical Society, 1992, 114(10): 3974-3975. DOI:10.1021/ja00036a053 |
| [11] |
SCHWAB P, GRUBBS R H, ZILLER J W. Synthesis and applications of RuCl2(CHR')(PR3)2: The influence of the alkylidene moiety on metathesis activity[J]. Journal of the American Chemical Society, 1996, 118(1): 100-110. DOI:10.1021/ja952676d |
| [12] |
RUFINO-FELIPE E, VALDÉS H, GERMÁN-ACACIO J M, et al. Fluorinated N-heterocyclic carbene complexes. applications in catalysis[J]. Journal of Organometallic Chemistry, 2020, 921: 121364. DOI:10.1016/j.jorganchem.2020.121364 |
| [13] |
SCHOLL M, DING S, LEE C W, et al. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1, 3-dimesityl-4, 5-dihydroimidazol-2-ylidene ligands[J]. Organic Letters, 1999, 1(6): 953-956. DOI:10.1021/ol990909q |
| [14] |
LOVE J A, MORGAN J P, TRNKA T M, et al. A practical and highly active ruthenium-based catalyst that effects the cross metathesis of acrylonitrile[J]. Angewandte Chemie (International Ed in English), 2002, 41(21): 4035-4037. DOI:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I |
| [15] |
CHOI T L, GRUBBS R H. Controlled living ring-opening-metathesis polymerization by a fast-initiating ruthenium catalyst[J]. Angewandte Chemie (International Ed in English), 2003, 42(15): 1743-1746. DOI:10.1002/anie.200250632 |
| [16] |
LEITGEB A, WAPPEL J, SLUGOVC C. The ROMP toolbox upgraded[J]. Polymer, 2010, 51(14): 2927-2946. DOI:10.1016/j.polymer.2010.05.002 |
| [17] |
SCHROCK R R. Synthesis of stereoregular polymers through ring-opening metathesis polymerization[J]. Accounts of Chemical Research, 2014, 47(8): 2457-2466. DOI:10.1021/ar500139s |
| [18] |
王晋宇, 柳春丽, 陈延辉. 胺基膦钌卡宾化合物的合成及催化烯烃复分解反应[J]. 高等学校化学学报, 2020, 41(12): 2766-2773. WANG Jinyu, LIU Chunli, CHEN Yanhui. Synthesis of aminophosphine ruthenium carbene complex and its application in olefins metathesis reaction[J]. Chemical Journal of Chinese Universities, 2020, 41(12): 2766-2773. (in Chinese) |
| [19] |
LYNN D M, GRUBBS R H. Novel reactivity of ruthenium alkylidenes in protic solvents: Degenerate alkylidene proton exchange[J]. Journal of the American Chemical Society, 2001, 123(14): 3187-3193. DOI:10.1021/ja0020299 |
| [20] |
STEWART I C, BENITEZ D, O'LEARY D J, et al. Conformations of N-heterocyclic carbene ligands in ruthenium complexes relevant to olefin metathesis[J]. Journal of the American Chemical Society, 2009, 131(5): 1931-1938. DOI:10.1021/ja8078913 |
| [21] |
CHU C, LIN T, SHAO H, et al. Disentangling ligand effects on metathesis catalyst activity: Experimental and computational studies of ruthenium-aminophosphine complexes[J]. Journal of the American Chemical Society, 2018, 140(16): 5634-5643. DOI:10.1021/jacs.8b02324 |
| [22] |
DUAN Y, WANG T, XIE Q, et al. Highly efficient nitrogen chelated ruthenium carbene metathesis catalysts[J]. Dalton Transactions, 2016, 45(48): 19441-19448. DOI:10.1039/C6DT03899A |
| [23] |
HONG S, WENZEL A G, SALGUERO T T, et al. Decomposition of ruthenium olefin metathesis catalysts[J]. Journal of the American Chemical Society, 2007, 129(25): 7961-7968. DOI:10.1021/ja0713577 |
| [24] |
RITTER T, HEJL A, WENZEL A G, et al. A standard system of characterization for olefin metathesis catalysts[J]. Organometallics, 2006, 25(24): 5740-5745. DOI:10.1021/om060520o |